製品・サービス情報
|
|
Abterra bio社 次世代シークエンシング(NGS)受託解析サービス
ReptorTM 免疫レパートリーシークエンス受託解析サービス |
|
 |
 抗体レパートリーの構築 抗体レパートリーの構築
|
免疫レパートリーは、特定時点で個人に存在する固有の免疫グロブリンおよびT細胞受容体配列の集合体です。Abterra bio社では、主に免疫グロブリン、つまりヒトおよび他の哺乳類のB細胞受容体レパートリーに焦点を当てています。成人ヒトにおける完全長免疫グロブリンをコードするB細胞および形質細胞の総数は、1010-1011と推定され、B細胞受容体または抗体レパートリーの総サイズに上限があります。抗体レパートリーは動的で、配列の多様性とレパートリーの組成は時間の経過とともに劇的に変化します。
高い配列多様性のため、抗体レパートリーシークエンシングと個々のレパートリーの組み立ては複雑であり、専用のツールが必要なため、標準的な RNA-seq
または標的濃縮ワークフローは適していません。抗体レパートリーのシークエンスと解析には、ユニークな課題があることを認識することが重要です。
◆抗体レパートリー構築の概要
|
【リード品質フィルタリング】
|
他のバイオインフォマティクスパイプラインと同様に、免疫レパートリー解析でも無意味なデータからは、無意味な結果がもたらされます。リードフィルタリングはプロセスの重要な部分であり、いずれの
RNA/DNA シークエンス解析においても最善の措置です。これには、低品質のリードの削除、スパイクインライブラリー(PhiXなど)から生成されたリードの削除、プライマーとシーケンスアダプターシークエンスのトリミングが含まれます。 |
【リードのつなぎ合わせ】
|
可変領域の330以上のヌクレオチド全体をカバーするには、ペアエンドリードまたは長いシングルエンドリードが必要です。ReptorTMサービスのワークフローでは、イルミナMiSeqシステムを使用して、2x300ヌクレオチドのペアリードを生成します。これにより、リード間でかなりの重複が発生します。オーバーラップを識別し、オーバーラップ領域にコンセンサス配列を構築することにより、ステッチリードを構築します。
|
|
 |
【長さと品質でフィルタリング】
|
リードペアが一つのコンセンサスリードとしてつなぎ合わされると、追加のフィルタリングが適用されます。具体的には、短すぎるまたは長すぎるコンセンサスリードを削除します。これは、リードが切り捨てられているか、つなげられていない可能性があります。ReptorTMワークフローでは、「抗体性」の大まかなテストも適用します。リードは、抗体遺伝子座特異的プライマーを使用した数ラウンドのPCR産物ですが、配列のサブセットは、オフターゲット遺伝子座である可能性があります。各配列が参照生殖細胞系列V遺伝子にほぼ一致するかどうかを迅速にテストします。各リードとリファレンス遺伝子の間に適切なアライメントが見つからない場合、リードは抗体ではない可能性があります(またはリファレンスが不完全です)。この段階では、同一の読み取り値を1つのリードに集約します。 |
【エラー修正】
|
1アミノ酸の変化は、抗体の安定性と親和性を劇的に変える可能性があります。そのため、エラー修正されたレパートリーを構築することは、抗体の発見からクローンの追跡まで、多くのダウンストリームアプリケーションによって重要な最初のステップです。PCR増幅とシークエンシングの両方でエラーが発生する可能性があり、これを修正する必要があります。簡単なアプローチは、アバンダンスの閾値を適用し、リード数が一定数未満の配列を削除することです。このアプローチの背後にある考え方は、エラーは稀であり、真の抗体配列は複数回出現する必要があるということです。存在量の少ない正しいシークエンスは削除され、レパートリーに関する実際の重要な情報が削除されます。
Unique molecular identifiers(UMIs: 分子バーコード)は、エラー修正し、各抗体遺伝子座のRNA量を正確に定量化するために人気が高まっています(Khan
et al. 2016, Turchaninova et al. 2016)。Abterra bio社では、UCSDで開発されたエラー修正を実行するためのHamming
クラスタリングアプローチに基づいています(Safonova et al. 2015)。個別の抗体配列ごとにグラフにノードを作成し、シークエンスが事前定義されたハミング距離内にある場合はノード間にエッジを作成します。原則として、誤ったシークエンスは、正しいシークエンスも含むサブグラフに表示されます。コンセンサス配列は、密なサブグラフを切り離すことによって作成されます。 |
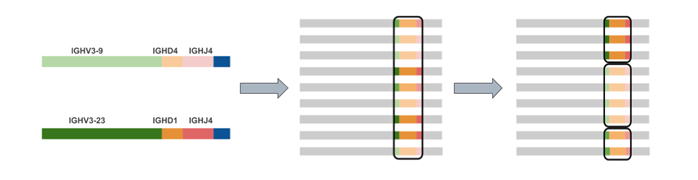
【V(D)Jラベリングと相補性決定領域の識別】
|
エラー修正後、レパートリーは修正されたシークエンスのコレクションで構成されます。レパートリーをさらに特徴づけるためには、各抗体を生じた生殖細胞系配列を決定する必要があります。このプロセスはV(D)Jラベリングと呼ばれ、IMGT/HighV-Quest、IgBLAST、MiXCRなどのオープンソースツールがこの目的のために開発されました。カラー抗体グラフ法(Bonissone
and Pevzner, 2016)を適用して、各抗体配列の生殖細胞系 V、D、 およびJ遺伝子を決定します。元の生殖細胞系配列を特定することに加えて、各抗体を生殖細胞系列にアライメントすることで、超変異部位を決定することができます。
生殖細胞系遺伝子の標識は、相補性決定領域(CDR)の同定にも役立ちます。各抗体鎖には、3つのCDRがあり、そのうちの2つは完全にV遺伝子セグメントに存在し、3つ目はV、D、およびJ(または軽鎖の場合はVとJ)の接合部に存在します。これらの領域を特定することは、レパートリー内の抗体間の関係、および免疫学的課題に応じたそれらの進化を理解するために重要です。私たちのプロセスでは、ほぼ同一のCDR3配列を単一のグループまたはクローンにクラスター化します。次の図は、V(D)Jラベリング、CDR3
識別、および CDR3 クラスタリングのプロセスを示しています。
|
|
|
 ヒトにおける免疫レパートリーシークエンシング解析例 ヒトにおける免疫レパートリーシークエンシング解析例
|
ReptorTMパイプラインを用いて、末梢血単核細胞(PBMC)由来のB細胞受容体(BCR)レパートリーを、異なる重鎖アイソタイプ(IgG、IgM、IgA、IgD、IgE)のレパートリー特性を調べました。
【シークエンシングと取得データ】
|
PBMCサンプルからRNAを抽出後、cDNAを構築し、cDNAを5サンプルに分けました。各サンプルから、独自のアイソタイプ特異的プライマーを用いて、5'RACE
ストラテジーにおいてBCRの可変領域を増幅しました。各ライブラリーを Illumina Miseq システムにて、1ライブラリーあたり 500K-1.5M
2x300bp リードの depth までシークエンシングしました。
Reptor パイプラインの前処理として、シークエンシングリードから非抗体汚染物質、低品質リード、および1つの配列につなぎ合わせられないペアエンドリードが除去されます。
MiSeqシステムエラー率は1%であり、全リードに少なくとも1つのエラーがあるとされています。エラー修正後、Reptorはエラー修正のための最小存在閾値(2リード)未満のリード、短すぎる/長すぎるリード、または生殖細胞系遺伝子セグメントにうまくアライメントできないリードをフィルタリングします。
| Library |
Preprocessed Reads |
Unique Antibodies(nt) |
Unique CDR3s(aa) |
| IgM |
568,474 |
31,292 |
27,971 |
| IgG |
605,251 |
11,452 |
7,463 |
| IgE |
149,727 |
171 |
26 |
| IgD |
504,814 |
11,068 |
8,190 |
| IgA |
857,386 |
20,257 |
11,544 |
|
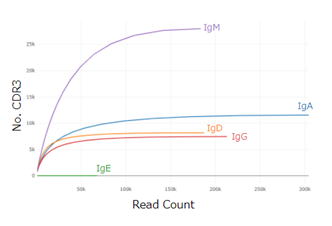
【IgM 発現抗体は他のアイソタイプよりも多様】
|
IgM アイソタイプは、ナイーブB細胞および一部のメモリーB細胞で発現します。当然のことながら、IgM抗体の中で最大の多様性が見られます。他のアイソタイプと比較して、IgM抗体は両方とも、固有の抗体配列の総数が多く、CDR3配列の数が最も多く、抗体あたりの平均リード数が最も少なくなっています。IgM抗体の45%は、生殖細胞系列と比較してV遺伝子セグメントに変異がありませんでした。
各アイソタイプのクローン多様性は、上記の希薄化曲線におけるシークエンスリードの経験的リサンプリング関数として測定されます。プロットはReptorによって自動的に生成され、様々なアイソタイプにわたるCDR3の多様性の比較を提供します。さらに、曲線が飽和しており、このライブラリーをより深くシークエンスしても、追加のCDR3は得られないことがわかります。IgMアイソタイプはCDR3で最大の多様性を示し、IgEは最小の多様性を示します。IgE発現抗体は最も多様性が低いです。IgE抗体は、特定の病原体に対するアレルギー反応と免疫応答に関与していますが、血清IgEの存在量とIgEを発現するB細胞の頻度の両方で、血液中ではまれであることが観察されています[1]。IgEレパートリーのサイズが小さいため、その後のレパートリー分析から除外されました。 |
|
 |
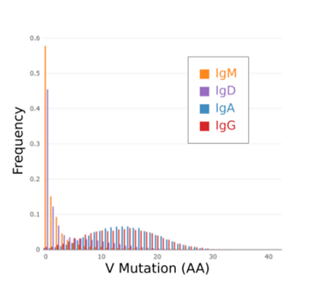
【変異分布はアイソタイプによって異なる】
|
各抗体について、ReptorはV遺伝子の生殖細胞系列に関して可変領域の変異を特定します。下図の標準的なReptor出力プロットは、抗体の頻度とアミノ酸変異数のヒストグラフを示しています。各アイソタイプは別系列として示され、レパートリーは主にナイーブ(IgD
及び IgM)または主に変異(IgG 及び IgA)のいずれかに分類できることがすぐに明らかになります。 |
|
 |
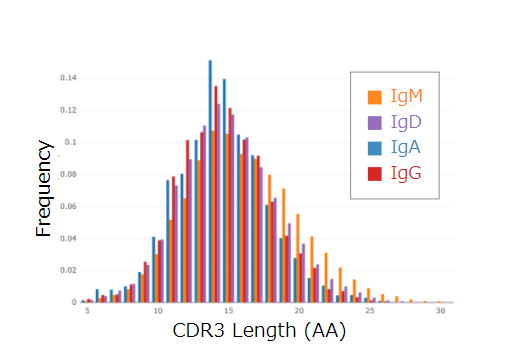
【CDR3 の長さはアイソタイプ間で一致】
|
アイソタイプ間の CDR3 の長さの比較は、レパートリー間に顕著な違いがないことを示しています。すべてのアイソタイプの様式は、14アミノ酸であり、これは他に報告されているレパートリーと一致しています[2]。 |
|
 |
【CDR3 の長さはアイソタイプ間で一致】
|
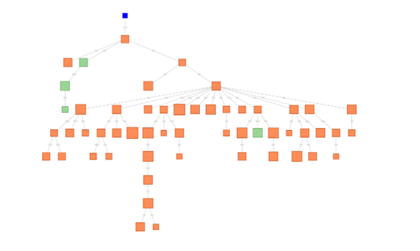
複数のアイソタイプで検出された CDR3 はごくわずかであり、PBMC由来BCRのバルクサンプリングでは予想外ではありません。共有クローンの最大の割合は
IgA と IgG の間で発生し、いずれかのレパートリーのクローンの3%(18,438 CDR3 のうち 569 CDRs)が両方のレパートリーで見つかりました。IgM
レパートリーは IgA(39,204 クローン中 311クローン)と IgD(35,891クローン中270クローン)の両方とクローンを共有しましたが、IgGと共有されたのは、35,328クローンのうち106クローンのみでした。
以下に編集距離の最小スパニングツリーは、同じ CDR3 配列を共有する異なる抗体配列間の関係を示しています。緑のボックスは、IgGレパートリーからのものであり、オレンジのボックスは
IgAレパートリーからのものです。青いノードは、この系統の理論上の(ただし観察されていない)生殖細胞系列の根です。 |
|
 |
【結論】
|
柔軟な免疫レパートリーシークエンステクノロジーである Reptor を使用して、健康なヒトのベースラインBCRレパートリーを分析しました。
分析から、アイソタイプ間の類似点と相違点を観察することができました。レプターは、次の様な幅広いアプリケーションに使用できます。
・時系列分析: Reptor は、ワクチンなどの免疫課題に応じて、時間の間に持続するクローンを特定します。
・空間分析: Reptor は、末梢血、リンパ節、骨髄などの組織全体の免疫レパートリーを比較します。
・症例制御分析: 複数の患者を Reptor によって同時に分析し、クラス間の違いを特定することができる。 |
【References】
|
 シークエンスエラー: 免疫レパートリーシークエンスにおけるエラー修正の重要性 シークエンスエラー: 免疫レパートリーシークエンスにおけるエラー修正の重要性
|
免疫レパートリーシークエンス(Rep-seq)を行うことで、個人のB細胞応答を構成する抗体配列の集団をデコンボリューションできます。抗体レパートリーの多様性は広大であることが知られていますが、エラーは真のバリアントを不明瞭にし、基礎となるサンプルについての結論を無効にします。エラーには主に2つの原因があります。
1)PCR増幅ステップによるエラー、2)シークエンス自体に起因するエラー
以下では、PCRに起因するエラーではなく、シークエンスに起因するエラーの影響のみを検討しました。 |
|
抗体レパートリーシークエンスは、エラー率が比較的低く、スループットが高く(1ランあたり約3000万リード)、抗体の可変領域をカバーするのに十分な長さのリード長(各300ntの重複リード)を満たすため、主にイルミナ
MiSeq シークエンサーを使用しています。ただし、高品質であるにも関わらず MiSeq の平均基本エラー率は1%(1)であり、主に置換エラーです。これらは抗体の通常の可変領域の約360nt
で3~4のエラーが予想されることを意味します。これは、MiSeqエラー率(例えば、位置依存性など)を単純化したものではありますが、有用です。
|
|
 |
|
各リードでエラーが発生する可能性があるため、文献では読み取りの処理に様々なアプローチが用いられています。
1.何もしない。エラーが少ないと仮定してすべてのリードを使用し、ユニークリードを集約。
2.ユニークなリードを集約、グローバルな閾値以上のリードを保持する(例:min abundance2)。
3.クラスタリングに基づいたエラー補正を行う(Abterra Bio のアプローチ、詳細は後述)。
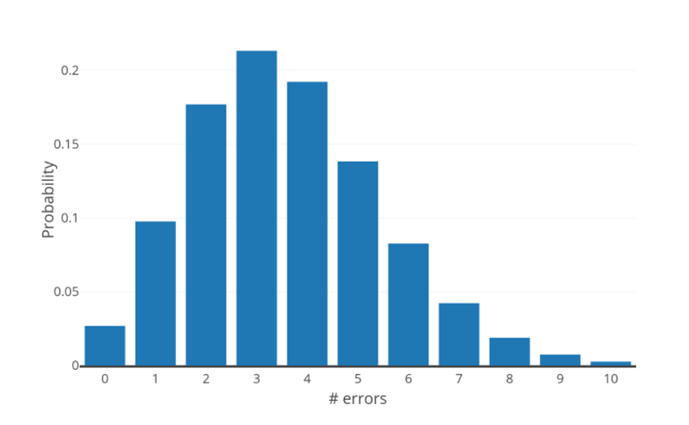
1番と2番の方法は似ている様に見えますが、2番の方法は最もシンプルなエラー補正と解釈できます。つまり、独立して複数回観測された配列は、同じ位置にランダムなエラーが発生する可能性が低く、正しい可能性が高いのです。残念ながら、2番の方法では、リードのごく一部しか残らないため、非常に無駄が多く、一方、1番の方法では、シークエンスアーティファクトではなく、真の多様性を特徴づけるための非常に現実的な問題を無視しています。
上の図は、360ntの配列に対して0.01の確率で発生するエラー予想分布を示しています(二項分布として計算されています)。
この分布は、なぜ1番目のアプローチが合理的でないかを示しています。
◆抗体レパートリーシークエンシングに対する誤差の影響
|
【エラー処理の一般的なアプローチ】
|
シークエンスエラーがコンパイルされたレパートリーの多様性に与える影響を示すために、レパートリーをシュミレートして(すなわち、V、D、Jの組み換えと体細胞超変異イベントをシュミレートして)、グランドトゥルースデータセットを得た。このレパートリーは、V、D,J遺伝子の使用量が一様で、配列ごとに
10+/-4nt の変異があるものとしてシュミレートされ、ARTリードシュミレーターを用いて、シュミレートされたリードシークエンシングを行った(2)。10,000のユニークなレパートリー配列から、各10の均一な深さをシュミレートした結果、Illumina
MiSeqのエラープロファイルで100,000シュミレートされたペアエンドリードが得られました。次に、これらのリードを Abterra bio社のReptorTMパイプラインで処理しました。
簡単に説明すると、ReptorTMは Hamming graph を用いてエラー補正を行い、類似したリードをクラスタリングします(3)。すべてのユニークなリードは、Hamming
graphHG=(V,E) のノードであり、HammingDist(u,v)<=tau の場合にのみ、Vの2つのノード u, v の間にエッジが引かれます。tau
パラメータは、HGがどの程度持続されるかを制御します。グラフが構築されると、密なサブグラフが特定され、リードの分割に使用されます。次に、パーテーション内のすべてのリードでコンセンサス配列を取り、エラーを除去します。
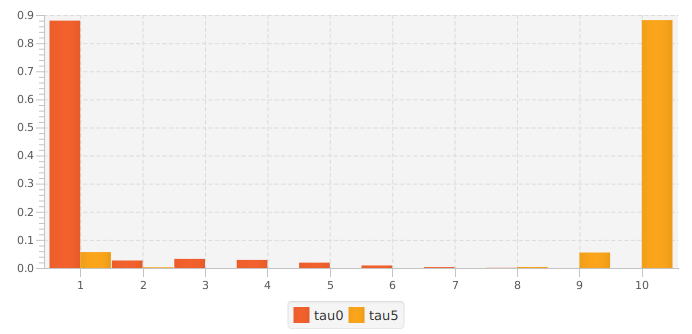
上記の3つのアプローチを考慮すると、次の様になります。
1. 68,639 のユニークリード。
2. 最小アバンダンス2を持つ 8,235 のユニークリード(入力リード全体のわずか12%)
3. 最小アバンダンス2を持つ 9,105 のユニークなクラスター(コンセンサスリード)(正しいアバンダンス10では 8,522)
上の図は、2つの方法(#1と#3)のカウント数の分布を示しています。何もしない場合、すべてのリードが残りますが、ほぼすべてが(間違って)シングルトンになっています。最小アバンダンスのカットオフを行うと、エラーの数は減りますが、リードの88%が捨てられています。方法#3(パラメータ
tau=5 を使用)では、全リードの94%を保持し、88.4%が正しいアバンダンス(アバンダンス10)である。 |
【クラスタリングの忠実度】
|
類似性に基づいてリードをクラスタリングすることでどの様にエラーが改善されるかを理解する1つの方法として、リードのパーティショニングの質を真のパーティショニングと比較して測定する方法があります。これらは、Jaccard
類似度や Fowlkes-Mallows index(FM) を用いて測定することができ、0.0の値は分離したパーティショニングを、1.0の値は正確なパーティショニングを表します。さらに、観測されたユニークな配列と期待されるユニークな配列の比率として定義されるインフレーション指数によって、外部の誤った多様性を測定することもできます。この比率が1.0の場合は完全な表現であり、1.0を超える場合は、多様性が増大していることを示しています。
下の表は、ReptorTM ハミング・グラフ・アプローチに対するパラメータ tau の影響を示しています。ここで、tau=0 は、ユニークリードのみを集約します(つまり、前のセクションのアプローチ#1)。この表から
tau==5 以降では、パーティションは変化しないことがわかります。これは、効果的な少数の追加リードがクラスター化され、追加の利益は実現しないことを示唆しています。また、この均一なデータセットでは、過剰な補正が行われていないことがわかります。
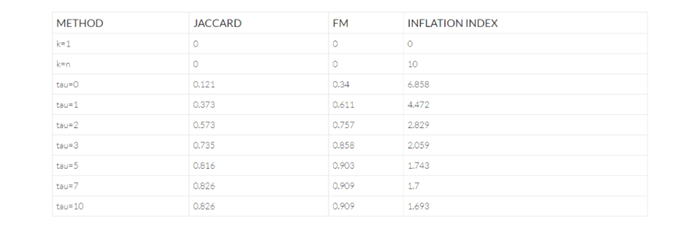
上記の表には、k=n (つまり、すべてのリードを独自のクラスターにクラスタリングする)と k=1 (つまり、すべてのリードを単一のクラスターにクラスタリングする)の2つのダミー行が含まれており、これらのエッジケースでの統計の効果を示しています。
|
【エラー修正のその他のアプローチ】
|
このブログ記事では、免疫系レパートリーのシークエンスエラーを処理することの重要性を強調しましたが、エラーを無視することは実行可能な選択肢ではありません。Reptorは、エラー修正に対して純粋にアルゴリズム的なアプローチをとっていますが、もう一つの一般的なアプローチは、ライブラリー作成工程でユニークな分子識別子(UMI)を組み込むことです。UMIは、ライブラリー作成時に、各分子に短い独自の識別用オリゴヌクレオチドを付加する必要があります。その後、このUMIに対してクラスタリングを行い、同じ分子からのリードのみをグループ化し、コンセンサスを取ってエラーを除去することができる。この方法は魅力的ですが、欠点もあります。
UMIは、衝突の可能性を減らすために、一様にランダムに生成する必要があります。しかし、長いランダムなヌクレオチドを一様な分布から合成するのは難しく、時にはスペーサーを組み込む必要があり、アンプリコンサイズが大きくなってしまう。さらに、2つの異なるUMIの間で二量体化が起こると、検出が困難なキメラ製品ができることがあります。最後に、PCRやシークエンスのエラーがUMIに蓄積される可能性があり、その場合、配列の消失を防ぐために、UMIの配列にクラスタリング(ハミンググラフを用いて)を行う必要があるかもしれません(4)。ハミンググラフによるエラー修正アプローチとUMIの完全な比較は、このブログ記事の範囲を超えていますが、いずれのアプローチも、レパートリーの人工的な多様性を無視するよりは望ましいことは確かです。 |
【References】
|
1. |
Melanie Schirmer, Rosalinda DAmore, Umer Z Ijaz, Neil Hall, and Christopher
Quince. Illumina error profile: resolving fine-scale variation in metagenomic
sequencing data. BMC bioinformatics, 17(1):125, 2016.(Pubmed: 26968756) |
| 2. |
Weichun Huang, Leping Li, Jason R Myers, and Gabor T Marth. ART: a next-generation
sequencing read simulator. Bioinformatics, 28(4): 593-594, 2011. (Pubmed:
22199392) |
| 3. |
Y. Safonova, S. Bonissone, E. Kurpilyansky, E. Starostina, A. Lapidus,
J. Stinson, L. DePalatis, W. Sandoval, J. Lill, and P. A. Pevzner. IgRepertoireConstructor:
a novel algorithm for antibody repertoire construction and immunoproteogenomics
analysis. Bioinformatics, 31(12):53-61, Jun 2015 (Pubmed: 26072509) |
| 4. |
Smith, Tom, Andreas Heger, and Ian Sudbery. "UMI-tools: modeling sequencing
errors in Unique Molecular Identifiers to improve quantification accuracy."
Genome research 27.3 (2017): 491-499. (Pubmed: 28100584) |
|
|
本ページは、Abterra bio社のホームページに掲載されている情報や画像を一部引用しています。
|
